the phydms_prepalignment program¶
Contents
Overview¶
phydms_prepalignment is a program that is useful for preparing your sequence alignment for input to phydms.
It can help you make codon-level alignments from protein alignments of translated coding sequences. It also performs basic checks on sequences, and can remove overly diverged or similar sequences. The basic notion is that you have some reference sequence (presumably what you used for the deep mutational scanning), and want to get homologs aligned to that for analysis.
See below for information on Command-line usage and Examples.
Command-line usage¶
Prepare alignment of protein-coding DNA sequences. Steps:
Any sequences specified by ‘–purgeseqs’ are removed.
Sequences not of length divisible by 3 are removed.
Sequences with ambiguous nucleotides are removed.
Sequences with non-terminal stop codons are removed; terminal stop codons are trimmed.
Sequences that do not encode unique proteins are removed unless they are specified for retention by ‘–keepseqs’.
A multiple sequence alignment is built using MAFFT. This step is skipped if you specify ‘–prealigned’.
Sites gapped in reference sequence are stripped.
Sequences with too little protein identity to reference sequence are removed, counting both mismatches and unstripped gaps as differences. Identity cutoff set by ‘–minidentity’.
Sequences too similar to other sequences are removed. An effort is made to keep one representative of sequences found many times in input set. Uniqueness threshold set by ‘–minuniqueness’. You can specify sequences to not remove via ‘–keepseqs’.
Problematic characters in header names are replaced by underscores. This is any space, comma, colon, semicolon parenthesis, bracket, single quote, or double quote.
An alignment is written, as well as a plot with same root but extension ‘.pdf’ that shows divergence from reference of all sequences retained and purged due to identity or uniqueness.
The ‘phydms’ package is written by the Bloom lab (see https://github.com/jbloomlab/phydms/contributors). Version 2.4.0 Full documentation at http://jbloomlab.github.io/phydms
usage: phydms_prepalignment [-h] [--prealigned] [--mafft MAFFT]
[--minidentity MINIDENTITY]
[--minuniqueness MINUNIQUENESS]
[--purgeseqs [PURGESEQS [PURGESEQS ...]]]
[--keepseqs [KEEPSEQS [KEEPSEQS ...]]] [-v]
inseqs alignment refseq
Positional Arguments¶
- inseqs
FASTA file giving input coding sequences.
- alignment
Name of created output FASTA alignment. PDF plot has same root, but extension ‘.pdf’.
- refseq
Reference sequence in ‘inseqs’: specify substring found ONLY in header for that sequence.
Named Arguments¶
- --prealigned
Sequences in ‘inseqs’ are already aligned, do NOT re-align.
Default: False
- --mafft
Path to MAFFT (http://mafft.cbrc.jp/alignment/software/).
Default: “mafft”
- --minidentity
Purge sequences with <= this protein identity to ‘refseq’.
Default: 0.7
- --minuniqueness
Require each sequence to have >= this many protein differences relative to other sequences.
Default: 2
- --purgeseqs
Specify sequences to always purge. Any sequences with any of the substrings specified here are always removed. The substrings can either be passed as repeated arguments here, or as the name of an existing file which has one substring per line.
- --keepseqs
Do not purge any of these sequences for lack of identity or uniqueness. Specified in the same fashion as for ‘–purgeseqs’.
- -v, --version
show program’s version number and exit
Examples¶
Let’s say we have some unaligned homologous protein-coding sequences in SUMO1_orthologs.fasta. Let’s say we want to use as our reference sequence the human ortholog, which has the header >ENSP00000376077_Hsap/1-303 (note the Hsap, indicating Homo sapiens). We can build a codon alignment with:
phydms_prepalignment SUMO1_orthologs.fasta SUMO1_alignment.fasta Hsap --minidentity 0.7 --minuniqueness 2
This creates the alignment SUMO1_alignment.fasta in which all gaps relative to the Homo sapiens orthologs are stripped, all sequences have at least 70% identity to the Homo sapiens ortholog, and all sequences have at least two protein differences from other retained homologs.
In addition to creating the alignment, the plot SUMO1_alignment.pdf is crated which shows the nucleotide and protein divergence from the reference Homo sapiens sequence of all homologs that passed the filters related to no premature stops, divisible by 3, and no stop codons. Here is that plot:
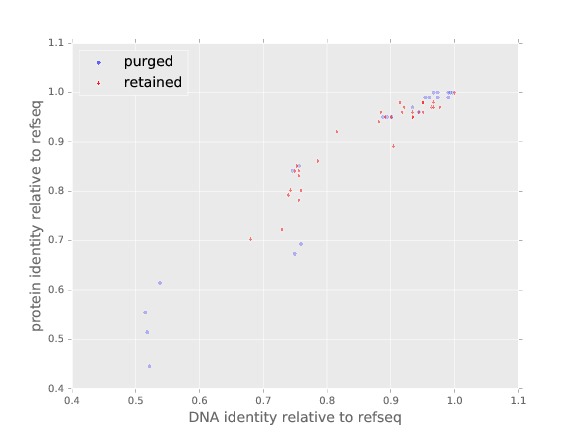
A jpg version of the plot SUMO1_alignment.pdf.¶
Sometimes there may be certain sequences you do not want to purge even if they lack uniqueness or identity to the reference sequence. For instance, maybe you want to keep the chimpanzee and gorilla homologs (which have Panu and Ggor in their headers) even though they are less than two amino-acid mutations from the human ortholog. In this case, use --keepseqs:
phydms_prepalignment SUMO1_orthologs.fasta SUMO1_alignment.fasta Hsap --minidentity 0.7 --minuniqueness 2 --keepseqs Panu Ggor
If there are many such sequences, you can also list them in a file, such as keepseqsfile.txt:
Panu
Ggor
and then specify this file to --keepseqs as:
phydms_prepalignment SUMO1_orthologs.fasta SUMO1_alignment.fasta Hsap --minidentity 0.7 --minuniqueness 2 --keepseqs keepseqsfile.txt
If there are sequences that you specifically want to exclude, you can do something similar with --purgseqs.


