Single-cell virus sequencing¶
This example shows how to use alignparse to process full-length PacBio CCSs of viral cDNAs generated in single-cell virus sequencing of influenza-infected cells. Specifically, it processes a snippet of data taking from Russell et al, 2019, which was generated by PacBio sequencing of viral cDNAs with unique molecular identifiers (UMIS) and cell barcodes added using a 10X Chromium (v2 reagents). This notebook aligns these CCSs to the influenza transcripts and parses the barcodes / UMIs and viral mutations.
Set up for analysis¶
Import necessary Python modules. We use alignparse for most of the operations, plotnine for ggplot2-like plotting:
[1]:
import os
import warnings
import numpy
import pandas as pd
from plotnine import *
import alignparse.ccs
import alignparse.consensus
import alignparse.minimap2
import alignparse.targets
from alignparse.constants import CBPALETTE
Suppress warnings that clutter output:
[2]:
warnings.simplefilter("ignore")
Directory for output:
[3]:
outdir = "./output_files/"
os.makedirs(outdir, exist_ok=True)
Target amplicons¶
The alignment targets consist of PCR amplicons covering the influenza virus mRNAs (WSN strain) with a 5’ termini corresponding to the primer binding site, the 3’ polyA tail used for reverse transcription, the UMI and cell barcode, and the common 3’ termini. There are also variant tags distinguishing the synonymously tagged viral variants (see Russell et al, 2019).
First, let’s look at the Genbank file holding the targets. The targets are defined in Genbank Flat File format Below we show the first 85 lines (the actual file is quite large as it defined all 10 influenza mRNAs):
[4]:
targetfile = "input_files/flu_WSN_amplicons.gb"
nlines_to_show = 85
with open(targetfile) as f:
print("".join(next(f) for _ in range(nlines_to_show)))
LOCUS fluPB2 2395 bp DNA UNK 01-JAN-1980
DEFINITION fluPB2.
ACCESSION fluPB2
VERSION fluPB2
KEYWORDS .
SOURCE .
ORGANISM .
.
FEATURES Location/Qualifiers
termini5 1..34
variant_tag_1 143..143
variant_tag_2 149..149
variant_tag_3 2163..2163
variant_tag_4 2171..2171
sequenced_mRNA 35..2316
final_mRNA_nts 2317..2319
polyA 2320..2347
UMI 2348..2357
cellbarcode 2358..2373
termini3 2374..2395
ORIGIN
1 gcgaaagcag gtcaattata ttcaatatgg aaagaataaa agaactaagg aatctaatgt
61 cgcagtctcg cactcgcgag atactcacaa aaaccaccgt ggaccatatg gccataatca
121 agaagtacac atcaggaaga cargagaara acccagcact taggatgaaa tggatgatgg
181 caatgaaata tccaattaca gcagacaaga ggataacgga aatgattcct gagagaaatg
241 agcagggaca aactttatgg agtaaaatga atgacgccgg atcagaccga gtgatggtat
301 cacctctggc tgtgacatgg tggaatagga atggaccagt gacaagtaca gttcattatc
361 caaaaatcta caaaacttat tttgaaaaag tcgaaaggtt aaaacatgga acctttggcc
421 ctgtccattt tagaaaccaa gtcaaaatac gtcgaagagt tgacataaat cctggtcatg
481 cagatctcag tgccaaagag gcacaggatg taatcatgga agttgttttc cctaacgaag
541 tgggagccag gatactaaca tcggaatcgc aactaacgac aaccaaagag aagaaagaag
601 aactccaggg ttgcaaaatt tctcctctga tggtggcata catgttggag agagaactgg
661 tccgcaaaac gagattcctc ccagtggctg gtggaacaag cagtgtgtac attgaagtgt
721 tgcatttgac ccaaggaaca tgctgggaac agatgtacac tccaggaggg gaggcgagga
781 atgatgatgt tgatcaaagc ttaattattg ctgctagaaa catagtaaga agagccacag
841 tatcagcaga tccactagca tctttattgg agatgtgcca cagcacgcag attggtggaa
901 taaggatggt aaacatcctt aggcagaacc caacagaaga gcaagccgtg gatatttgca
961 aggctgcaat gggactgaga attagctcat ccttcagttt tggtggattc acatttaaga
1021 gaacaagcgg atcatcagtc aagagagagg aagaggtgct tacgggcaat cttcagacat
1081 tgaagataag agtgcatgag ggatatgaag agttcacaat ggttgggaga agagcaacag
1141 ctatactcag aaaagcaacc aggagattga ttcagctgat agtgagtggg agagacgaac
1201 agtcgattgc cgaagcaata attgtggcca tggtattttc acaagaggat tgtatgataa
1261 aagcagttag aggtgacctg aatttcgtca atagggcgaa tcagcgattg aatcccatgc
1321 accaactttt gagacatttt cagaaggatg caaaggtgct ctttcaaaat tggggaattg
1381 aatccatcga caatgtgatg ggaatgatcg ggatattgcc cgacatgact ccaagcaccg
1441 agatgtcaat gagaggagtg agaatcagca aaatgggggt agatgagtat tccagcgcgg
1501 agaagatagt ggtgagcatt gaccgttttt tgagagttag ggaccaacgt gggaatgtac
1561 tactgtctcc cgaggagatc agtgaaacac agggaacaga gaaactgaca ataacttact
1621 catcgtcaat gatgtgggag attaatggtc ctgaatcagt gttggtcaat acctatcagt
1681 ggatcatcag aaactgggaa actgttaaaa ttcagtggtc ccagaatcct acaatgctgt
1741 acaataaaat ggaatttgag ccatttcagt ctttagttcc aaaggccgtt agaggccaat
1801 acagtgggtt tgtgagaact ctgttccaac aaatgaggga tgtgcttggg acatttgata
1861 ccgctcagat aataaaactt cttcccttcg cagccgctcc accaaagcaa agtagaacgc
1921 agttctcctc attgactata aatgtgaggg gatcaggaat gagaatactt gtaaggggca
1981 attctccagt attcaactac aacaagacca ctaaaagact cacagttctc ggaaaggatg
2041 ctggcccttt aactgaagac ccagatgaag gcacagctgg agttgagtcc gcagttctga
2101 gaggattcct cattctgggc aaagaagaca ggagatatgg accagcatta agcataaatg
2161 aaytgagcaa ycttgcgaaa ggagagaagg ctaatgtgct aattgggcaa ggagacgtgg
2221 tgttggtaat gaaacggaaa cggaactcta gcatacttac tgacagccag acagcgacca
2281 aaagaattcg gatggccatc aattagtgtc gaatagttta aaaaaaaaaa aaaaaaaaaa
2341 aaaaaaannn nnnnnnnnnn nnnnnnnnnn nnnagatcgg aagagcgtcg tgtag
//
LOCUS fluPB1 2395 bp DNA UNK 01-JAN-1980
DEFINITION fluPB1.
ACCESSION fluPB1
VERSION fluPB1
KEYWORDS .
SOURCE .
ORGANISM .
.
FEATURES Location/Qualifiers
termini5 1..22
variant_tag_1 146..146
variant_tag_2 152..152
variant_tag_3 2159..2159
variant_tag_4 2162..2162
sequenced_mRNA 23..2316
final_mRNA_nts 2317..2319
polyA 2320..2347
UMI 2348..2357
cellbarcode 2358..2373
termini3 2374..2395
ORIGIN
1 gcgaaagcag gcaaaccatt tgaatggatg tcaatccgac tttacttttc ttaaaagtgc
61 cagcacaaaa tgctataagc acaactttcc cttatactgg agaccctcct tacagccatg
We also have the YAML file specifying how to parse the features in the alignments. Note how this file uses the YAML syntax for defaults to avoid repeating the same specifications for each target:
[5]:
feature_parse_specs_file = "input_files/flu_WSN_feature_parse_specs.yaml"
with open(feature_parse_specs_file) as f:
print(f.read())
# default for genes with two variant tags
default_2tags: &default_2tags
query_clip5: 4
query_clip3: 4
termini5:
filter:
clip5: 4
mutation_nt_count: 1
mutation_op_count: null
sequenced_mRNA:
filter:
mutation_nt_count: null
mutation_op_count: 10
return: [mutations, accuracy]
final_mRNA_nts:
filter:
mutation_nt_count: null
mutation_op_count: 2
polyA:
filter:
mutation_nt_count: 12
mutation_op_count: 4
UMI:
return: [sequence, accuracy]
cellbarcode:
return: [sequence, accuracy]
termini3:
filter:
clip5: 4
mutation_nt_count: 1
mutation_op_count: null
variant_tag_1:
filter:
mutation_nt_count: 1
mutation_op_count: null
return: sequence
variant_tag_2:
filter:
mutation_nt_count: 1
mutation_op_count: null
return: sequence
# default for genes with four variant tags
default_4tags: &default_4tags
<<: *default_2tags
variant_tag_3:
filter:
mutation_nt_count: 1
mutation_op_count: null
return: sequence
variant_tag_4:
filter:
mutation_nt_count: 1
mutation_op_count: null
return: sequence
# use defaults to define specs for actual genes
fluPB2:
<<: *default_4tags
fluPB1:
<<: *default_4tags
fluPA:
<<: *default_4tags
fluHA:
<<: *default_4tags
fluNP:
<<: *default_4tags
fluNA:
<<: *default_4tags
fluM1:
<<: *default_4tags
fluM2:
<<: *default_2tags
fluNS1:
<<: *default_4tags
fluNS2:
<<: *default_2tags
We now read the targets into an alignparse.targets.Targets object with the feature-parsing specs (ignoring the keys that define thet defaults):
[6]:
targets = alignparse.targets.Targets(
seqsfile=targetfile,
feature_parse_specs=feature_parse_specs_file,
ignore_feature_parse_specs_keys=["default_2tags", "default_4tags"],
allow_extra_features=True,
)
Plot the Targets:
[7]:
_ = targets.plot(ax_width=10)
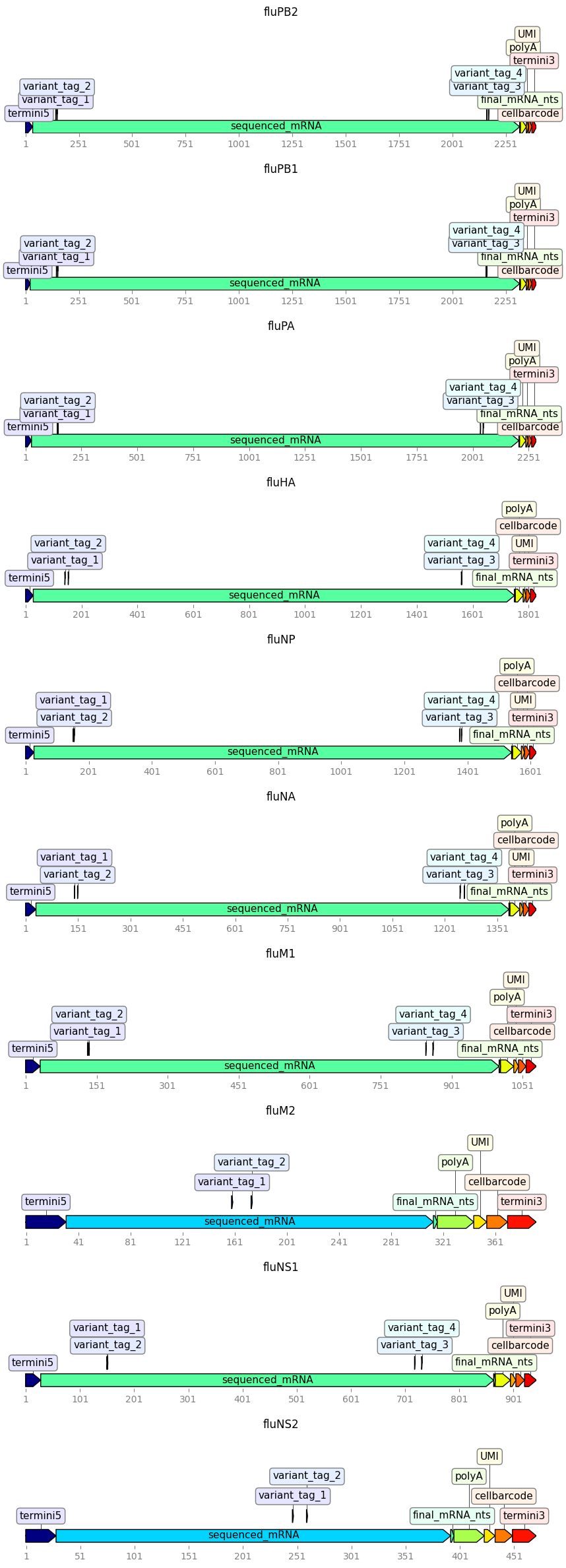
Several things of note about how these targets are defined. These all relate to the fact that the most difficult region to parse is the polyA tail and the UMI / cell barcode–the reason being that the polyA tail is not sequenced very accurately (it is a homopolymer) plus appears to be somewhat impure in the actual primers, so is difficult to align correctly. This is compounded by the fact that we don’t know the actual UMI sequence and so don’t want indels of the polyA tail to “bleed” into the UMI or gene alignment. Therefore:
We define a separate feature for the final nucleotides (last 3) of the mRNA upstream of the sequenced mRNA, so polyA indels don’t get assigned to the sequenced mRNA too often.
We define the polyA to be just 28 nucleotides in the alignment even though the length in the primer is 30, so that deletions don’t affect the UMI.
These empirically help with the alilgnments.
PacBio CCSs¶
Now let’s look at the PacBio CCSs. First, define a data frame with the information on the PacBio runs (here we just have one):
[8]:
pacbio_runs = pd.DataFrame(
{"name": ["flu_WSN"], "fastq": ["input_files/flu_WSN_ccs.fastq"]}
)
pacbio_runs
[8]:
| name | fastq | |
|---|---|---|
| 0 | flu_WSN | input_files/flu_WSN_ccs.fastq |
Create an alignparse.ccs.Summaries object:
[9]:
ccs_summaries = alignparse.ccs.Summaries(pacbio_runs, report_col=None)
Statistics on the CCSs (length, number of subread passes, quality):
[10]:
# NBVAL_IGNORE_OUTPUT
for stat in ["length", "passes", "accuracy"]:
if ccs_summaries.has_stat(stat):
p = ccs_summaries.plot_ccs_stats(stat)
p = p + theme(panel_grid_major_x=element_blank()) # no vertical grid lines
_ = p.draw()
else:
print(f"No {stat} statistics available.")
Align and parse¶
First, we create an alignparse.minimap2.Mapper to run minimap2, which is used for the alignments. We use minimap2 options that are tailored for viruses with potentially large internal deletions (which are handled like spliced introns); these options are specified by alignparse.minimap2.OPTIONS_VIRUS_W_DEL:
[11]:
# NBVAL_IGNORE_OUTPUT
mapper = alignparse.minimap2.Mapper(alignparse.minimap2.OPTIONS_VIRUS_W_DEL)
print(
f"Using `minimap2` {mapper.version} with these options:\n"
+ " ".join(mapper.options)
)
Using `minimap2` 2.22-r1101 with these options:
-xsplice:hq -un -C0 --splice-flank=no -M=1 --end-seed-pen=2 --end-bonus=2 --secondary=no --cs
Now use Targets.align_and_parse to align and parse the CCSs. First, create the output directory:
[12]:
align_and_parse_outdir = os.path.join(outdir, "flu_WSN_align_and_parse")
Now do the alignments and parsing:
[13]:
readstats, aligned, filtered = targets.align_and_parse(
df=pacbio_runs,
mapper=mapper,
outdir=align_and_parse_outdir,
name_col="name",
queryfile_col="fastq",
overwrite=True, # overwrite any existing output
ncpus=-1, # use all available CPUs
)
First, let’s look at the read alignment stats:
[14]:
readstats
[14]:
| name | category | count | |
|---|---|---|---|
| 0 | flu_WSN | filtered fluPB2 | 15 |
| 1 | flu_WSN | aligned fluPB2 | 50 |
| 2 | flu_WSN | filtered fluPB1 | 34 |
| 3 | flu_WSN | aligned fluPB1 | 165 |
| 4 | flu_WSN | filtered fluPA | 36 |
| 5 | flu_WSN | aligned fluPA | 173 |
| 6 | flu_WSN | filtered fluHA | 2 |
| 7 | flu_WSN | aligned fluHA | 8 |
| 8 | flu_WSN | filtered fluNP | 6 |
| 9 | flu_WSN | aligned fluNP | 23 |
| 10 | flu_WSN | filtered fluNA | 3 |
| 11 | flu_WSN | aligned fluNA | 18 |
| 12 | flu_WSN | filtered fluM1 | 7 |
| 13 | flu_WSN | aligned fluM1 | 14 |
| 14 | flu_WSN | filtered fluM2 | 1 |
| 15 | flu_WSN | aligned fluM2 | 2 |
| 16 | flu_WSN | filtered fluNS1 | 3 |
| 17 | flu_WSN | aligned fluNS1 | 20 |
| 18 | flu_WSN | filtered fluNS2 | 0 |
| 19 | flu_WSN | aligned fluNS2 | 0 |
| 20 | flu_WSN | unmapped | 108 |
Plot these stats:
[15]:
# NBVAL_IGNORE_OUTPUT
p = (
ggplot(
readstats.assign(
category=lambda x: pd.Categorical(
x["category"], x["category"].unique(), ordered=True
),
is_aligned=lambda x: x["category"].str.contains("aligned"),
),
aes("category", "count", fill="is_aligned"),
)
+ geom_bar(stat="identity")
+ facet_wrap("~ name", nrow=1)
+ theme(
axis_text_x=element_text(angle=90),
figure_size=(0.3 * len(readstats), 2.5),
panel_grid_major_x=element_blank(), # no vertical grid lines
)
+ scale_fill_manual(values=CBPALETTE)
)
_ = p.draw()
Now let’s look at the reason that reads that were filtered failed to align. Recall that filtered holds a data frame for each target:
[16]:
for target in targets.target_names[:1]:
print(f"First few lines of `filtered` for {target}:")
display(filtered[target].head())
First few lines of `filtered` for fluPB2:
| name | query_name | filter_reason | |
|---|---|---|---|
| 0 | flu_WSN | m54228_171201_172448/5308608/ccs | termini5 clip5 |
| 1 | flu_WSN | m54228_171201_172448/6554482/ccs | polyA mutation_nt_count |
| 2 | flu_WSN | m54228_171201_172448/7143925/ccs | polyA mutation_nt_count |
| 3 | flu_WSN | m54228_171201_172448/9437481/ccs | final_mRNA_nts mutation_op_count |
| 4 | flu_WSN | m54228_171201_172448/9830886/ccs | polyA mutation_nt_count |
Concatenate the information for all of the targets and plot reasons why mapped reads were filtered:
[17]:
# NBVAL_IGNORE_OUTPUT
p = (
ggplot(
pd.concat([df.assign(gene=gene) for gene, df in filtered.items()]).assign(
gene=lambda x: pd.Categorical(x["gene"], x["gene"].unique(), ordered=True)
),
aes("filter_reason"),
)
+ geom_bar()
+ facet_wrap("~ gene", ncol=5)
+ theme(
axis_text_x=element_text(angle=90),
figure_size=(9, 4),
panel_grid_major_x=element_blank(), # no vertical grid lines
)
)
_ = p.draw()
Now let’s look at the first few aligned reads for the first few genes:
[18]:
for target in targets.target_names[:4]:
print(f"\nFirst few entries in `aligned` for {target}:")
display(aligned[target].head())
First few entries in `aligned` for fluPB2:
| name | query_name | query_clip5 | query_clip3 | sequenced_mRNA_mutations | sequenced_mRNA_accuracy | UMI_sequence | UMI_accuracy | cellbarcode_sequence | cellbarcode_accuracy | variant_tag_1_sequence | variant_tag_2_sequence | variant_tag_3_sequence | variant_tag_4_sequence | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | flu_WSN | m54228_171201_172448/5243086/ccs | 0 | 0 | del279to1904 C2077A C2157A C2214A | 1.0 | CAATAGGTAC | 1.0 | AGTAGTCACTTCAAGC | 1.0 | G | G | C | C |
| 1 | flu_WSN | m54228_171201_172448/5833039/ccs | 0 | 0 | del117to2070 | 1.0 | AGCGCTCTTT | 1.0 | ATAAGGTGACCTCTCA | 1.0 | A | A | T | T |
| 2 | flu_WSN | m54228_171201_172448/5898400/ccs | 0 | 0 | G67T del152to1989 | 1.0 | TGTCACTGGC | 1.0 | TCGGAGCTGGTCTTAG | 1.0 | G | G | C | C |
| 3 | flu_WSN | m54228_171201_172448/6160599/ccs | 0 | 0 | G242T C430G del551to2282 | 1.0 | CTCGGTCCGC | 1.0 | GTGCGCATGGAAATCA | 1.0 | G | G | ||
| 4 | flu_WSN | m54228_171201_172448/6161088/ccs | 0 | 0 | del152to1989 G2057T G2118T | 1.0 | GGTTGGCGCT | 1.0 | CGGAGCTGGTCTTTAG | 1.0 | G | G | C | C |
First few entries in `aligned` for fluPB1:
| name | query_name | query_clip5 | query_clip3 | sequenced_mRNA_mutations | sequenced_mRNA_accuracy | UMI_sequence | UMI_accuracy | cellbarcode_sequence | cellbarcode_accuracy | variant_tag_1_sequence | variant_tag_2_sequence | variant_tag_3_sequence | variant_tag_4_sequence | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | flu_WSN | m54228_171201_172448/4784702/ccs | 0 | 0 | del171to2016 del2268to2268 | 1.000000 | GCGTATCCTG | 1.000000 | CCTTCGTGATGATTGC | 1.000000 | T | C | C | T |
| 1 | flu_WSN | m54228_171201_172448/4785077/ccs | 0 | 0 | del180to2032 | 1.000000 | GTGCATGTGT | 1.000000 | TTCAGCCTGTTCAAGC | 1.000000 | T | C | C | T |
| 2 | flu_WSN | m54228_171201_172448/4915675/ccs | 0 | 0 | del171to2016 del2070to2070 | 1.000000 | CACAAACTAT | 1.000000 | GCACAGCTGACTGATC | 1.000000 | T | C | C | T |
| 3 | flu_WSN | m54228_171201_172448/5111911/ccs | 0 | 0 | ins397T C468T T1612C | 0.999384 | AGCGTGCCAC | 0.999999 | ATCCTGGCTGCGTCCA | 0.999998 | T | C | C | T |
| 4 | flu_WSN | m54228_171201_172448/5112319/ccs | 0 | 0 | del258to2001 | 1.000000 | CAACGATGAC | 1.000000 | GGCCACAACGCGAATG | 1.000000 | T | C | C | T |
First few entries in `aligned` for fluPA:
| name | query_name | query_clip5 | query_clip3 | sequenced_mRNA_mutations | sequenced_mRNA_accuracy | UMI_sequence | UMI_accuracy | cellbarcode_sequence | cellbarcode_accuracy | variant_tag_1_sequence | variant_tag_2_sequence | variant_tag_3_sequence | variant_tag_4_sequence | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | flu_WSN | m54228_171201_172448/4587763/ccs | 0 | 0 | del117to1927 C2159A | 1.000000 | AAACTAGTGG | 1.0 | AAGACATGATCAGTGT | 1.0 | T | T | C | |
| 1 | flu_WSN | m54228_171201_172448/4653834/ccs | 0 | 0 | del179to1839 | 1.000000 | AGCACACATA | 1.0 | TCCCATTGAAAACACA | 1.0 | C | T | A | T |
| 2 | flu_WSN | m54228_171201_172448/4719236/ccs | 0 | 0 | C42A del179to1839 | 1.000000 | ACTGAAACAA | 1.0 | TCCCGTTGAAAACACA | 1.0 | C | T | A | T |
| 3 | flu_WSN | m54228_171201_172448/4784775/ccs | 0 | 0 | del66to177 del265to1996 C2028T | 0.999981 | AATAGTTCCC | 1.0 | GGCAAACGAGACGCAA | 1.0 | A | T | ||
| 4 | flu_WSN | m54228_171201_172448/4850043/ccs | 0 | 0 | A64G ins402A del1063to1067 del1073to2180 | 0.999704 | CATCATATTG | 1.0 | TGTGGTCCTACTGATC | 1.0 | T | C |
First few entries in `aligned` for fluHA:
| name | query_name | query_clip5 | query_clip3 | sequenced_mRNA_mutations | sequenced_mRNA_accuracy | UMI_sequence | UMI_accuracy | cellbarcode_sequence | cellbarcode_accuracy | variant_tag_1_sequence | variant_tag_2_sequence | variant_tag_3_sequence | variant_tag_4_sequence | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | flu_WSN | m54228_171201_172448/4522378/ccs | 0 | 0 | G397A ins1145A del1251to1722 | 1.000000 | TAGACCCTTT | 1.00000 | GAGTACACTATCGCGC | 1.0 | C | G | ||
| 1 | flu_WSN | m54228_171201_172448/6947580/ccs | 0 | 0 | del108to1459 | 1.000000 | CTTTGGGGGG | 0.99998 | GTAGGTTTGTCACTAG | 1.0 | G | T | ||
| 2 | flu_WSN | m54228_171201_172448/7144230/ccs | 2 | 0 | C260A C262A ins288G del471to471 C1052A ins1450T | 0.999627 | CGTACGGCAG | 1.00000 | AAACTGGCTAGGAGCT | 1.0 | A | A | A | C |
| 3 | flu_WSN | m54228_171201_172448/7340424/ccs | 0 | 0 | G213A ins663A G1715A del1718to1722 | 0.999847 | GCTTGCCGGC | 1.00000 | GGTCACTCTGCGTTAT | 1.0 | A | A | A | C |
| 4 | flu_WSN | m54228_171201_172448/8520141/ccs | 0 | 0 | G9T C153A del771to771 A1253C del1256to1722 | 0.999810 | GTGCCAAGGA | 1.00000 | GAAAGGCTGGTACGAG | 1.0 | C | G |

