Barcoded-subamplicon sequencing¶
Overview¶
Barcoded-subamplicon sequencing is a strategy to increase the accuracy of Illumina deep sequencing. This strategy is useful for deep mutational scanning, since it enables better identification of rare mutations without the confounding effects of sequencing errors.
The dms_tools2 software contains programs to process the deep-sequencing data generated by this sequencing strategy. Typically you would want to analyze your data using dms2_batch_bcsubamp, which in turn calls dms2_bcsubamp. The Examples include some analyses of this type of deep-sequencing data; for instance see the Doud2016 example.
How does barcoded-subamplicon sequencing work?¶
Barcoded-subamplicon sequencing is an Illumina library preparation strategy that increases the sequencing accuracy by attaching unique random barcodes to molecules during library preparation, and then grouping sequencing reads by barcode to correct for sequencing errors. To our knowledge, this basic concept was first described by Hiatt et al (2010), Kinde et al (2011), and Jabara et al (2011). The concept was first applied for use in deep mutational scanning by Wu et al (2014). Zhang et al (2016) reported that barcoded-subamplicon sequencing is a better error-correction strategy than overlapping paired-end reads, and our results in the Bloom lab are consistent with their report. The exact implementation of barcoded-subamplicon sequencing used by the Bloom lab (and the implementation that dms_tools2 is designed to analyze) is described in Doud and Bloom (2016).
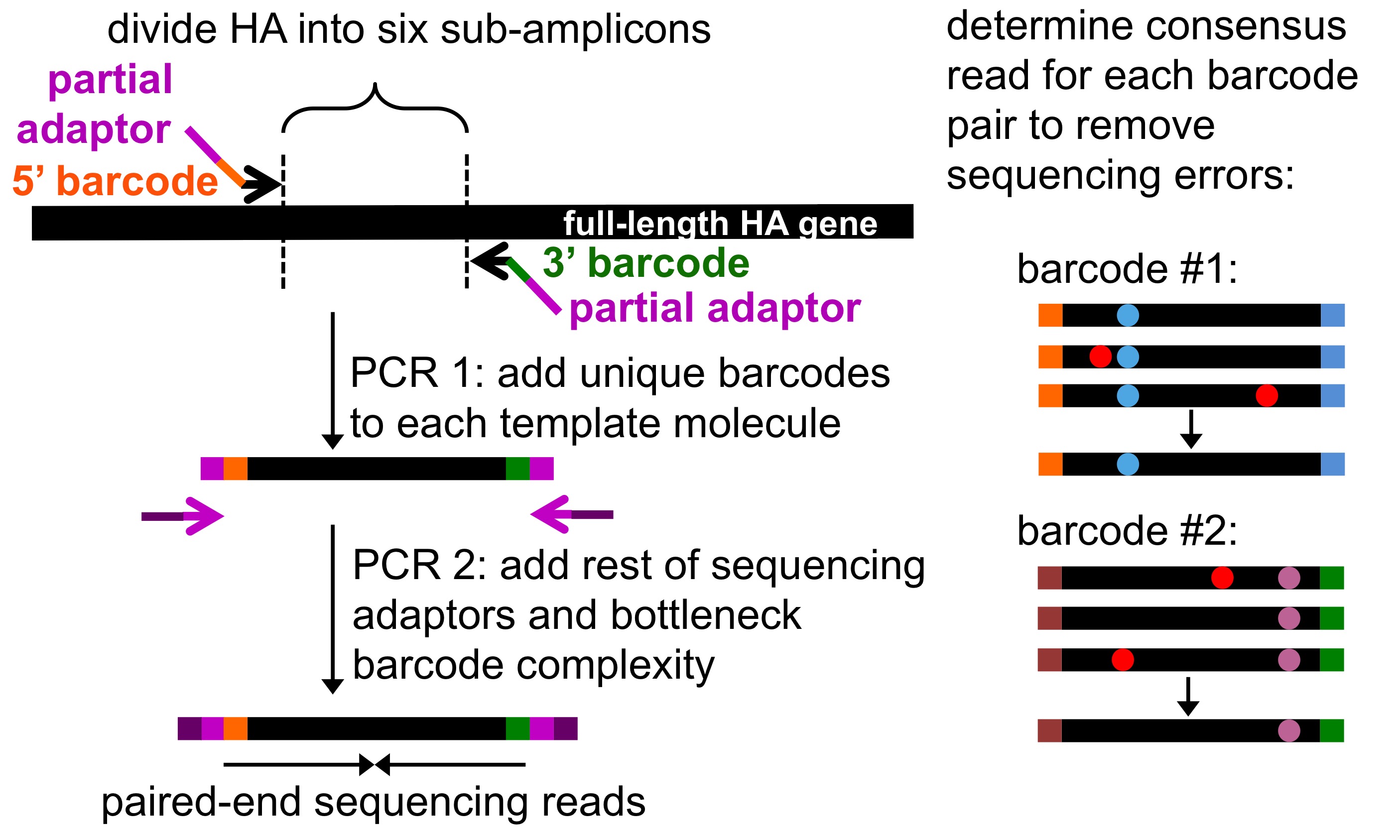
Fig. 1 Schematic of barcoded-subamplicon sequencing of influenza HA as done by Doud and Bloom (2016).¶
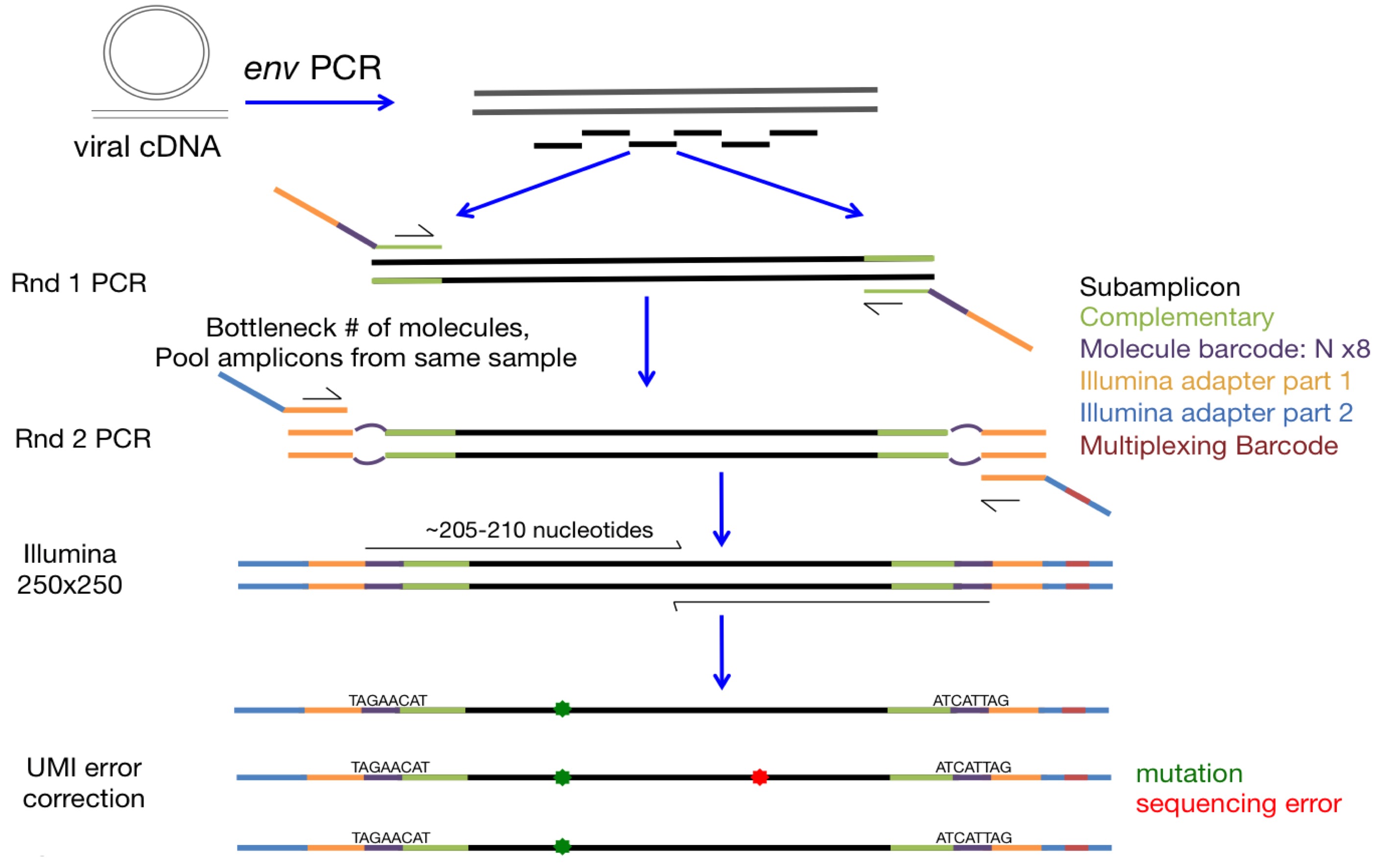
Fig. 2 Schematic of barcoded-subamplicon sequencing of HIV Env as done by Dingens et al (2017).¶
Schematics of barcoded-subamplicon sequencing are shown in Fig. 1 and Fig. 2.
Essentially, barcoded-subamplicon sequencing involves using PCR to make a series of subamplicons from the gene to be sequenced.
Each of these subamplicons contains a random barcode (a string of N nucleotides) at each end that provides a unique identifier.
The barcoded subamplicons are then diluted such that the number of unique ssDNA molecules is less than the read depth, and then this dilution is amplified by primers that also add the Illumina adaptors.
Finally, these molecules are sequenced to a depth such that most barcodes are observed multiple times with different reads.
In analyzing the sequencing, we identify barcodes that are sequenced multiple times, and build a consensus sequence for the subamplicon from these multiple reads. We only call mutations if multiple reads at a site all agree, thereby ameliorating the effect of sequencing errors that affect a single read. We then align each consensus subamplicon to the reference sequence to call mutations. There are more details in the Algorithm for assembling and aligning subamplicons described in the documentation for dms2_bcsubamp.
Workflow¶
To understand in detail how barcoded-subamplicon sequencing is done, example primers are shown below for sequencing of the hemagglutinin (HA) from A/WSN/1933 (H1N1) influenza:
Rnd1for1: 5'-CTTTCCCTACACGACGCTCTTCCGATCTNNNNNNNNaagcaggggaaaataaaaacaaccaaa-3' Rnd1for427: 5'-CTTTCCCTACACGACGCTCTTCCGATCTNNNNNNNNccaaggaaagttcatggcccaac-3' Rnd1for850: 5'-CTTTCCCTACACGACGCTCTTCCGATCTNNNNNNNNgtttgagtccggcatcatcacc-3' Rnd1for1276: 5'-CTTTCCCTACACGACGCTCTTCCGATCTNNNNNNNNcaacaacttagaaaaaaggatggaaaatttaaataaa-3'
WSN HA with coding sequence in caps: 5'-agcaaaagcaggggaaaataaaaacaaccaaaATGAAGGCAAAACTACTGGTCCTGTTATATGCATTTGTAGCTACAGATGCAGACACAATATGTATAGGCTACCATGCGAACAACTCAACCGACACTGTTGACACAATACTCGAGAAGAATGTGGCAGTGACACATTCTGTTAACCTGCTCGAAGACAGCCACAACGGGAAACTATGTAAATTAAAAGGAATAGCCCCACTACAATTGGGGAAATGTAACATCACCGGATGGCTCTTGGGAAATCCAGAATGCGACTCACTGCTTCCAGCGAGATCATGGTCCTACATTGTAGAAACACCAAACTCTGAGAATGGAGCATGTTATCCAGGAGATCTCATCGACTATGAGGAACTGAGGGAGCAATTGAGCTCAGTATCATCATTAGAAAGATTCGAAATATTTCCCAAGGAAAGTTCATGGCCCAACCACACATTCAACGGAGTAACAGTATCATGCTCCCATAGGGGAAAAAGCAGTTTTTACAGAAATTTGCTATGGCTGACGAAGAAGGGGGATTCATACCCAAAGCTGACCAATTCCTATGTGAACAATAAAGGGAAAGAAGTCCTTGTACTATGGGGTGTTCATCACCCGTCTAGCAGTGATGAGCAACAGAGTCTCTATAGTAATGGAAATGCTTATGTCTCTGTAGCGTCTTCAAATTATAACAGGAGATTCACCCCGGAAATAGCTGCAAGGCCCAAAGTAAGAGATCAACATGGGAGGATGAACTATTACTGGACCTTGCTAGAACCCGGAGACACAATAATATTTGAGGCAACTGGTAATCTAATAGCACCATGGTATGCTTTCGCACTGAGTAGAGGGTTTGAGTCCGGCATCATCACCTCAAACGCGTCAATGCATGAGTGTAACACGAAGTGTCAAACACCCCAGGGAGCTATAAACAGCAATCTCCCTTTCCAGAATATACACCCAGTCACAATAGGAGAGTGCCCAAAATATGTCAGGAGTACCAAATTGAGGATGGTTACAGGACTAAGAAACATCCCATCCATTCAATACAGAGGTCTATTTGGAGCCATTGCTGGTTTTATTGAGGGGGGATGGACTGGAATGATAGATGGATGGTATGGTTATCATCATCAGAATGAACAGGGATCAGGCTATGCAGCGGATCAAAAAAGCACACAAAATGCCATTAACGGGATTACAAACAAGGTGAACTCTGTTATCGAGAAAATGAACACTCAATTCACAGCTGTGGGTAAAGAATTCAACAACTTAGAAAAAAGGATGGAAAATTTAAATAAAAAAGTTGATGATGGGTTTCTGGACATTTGGACATATAATGCAGAATTGTTAGTTCTACTGGAAAATGAAAGGACTTTGGATTTCCATGACTTAAATGTGAAGAATCTGTACGAGAAAGTAAAAAGCCAATTAAAGAATAATGCCAAAGAAATCGGAAATGGGTGTTTTGAGTTCTACCACAAGTGTGACAATGAATGCATGGAAAGTGTAAGAAATGGGACTTATGATTATCCAAAATATTCAGAAGAATCAAAGTTGAACAGGGAAAAGATAGATGGAGTGAAATTGGAATCAATGGGGGTGTATCAGATTCTGGCGATCTACTCAACTGTCGCCAGTTCACTGGTGCTTTTGGTCTCCCTGGGGGCAATCAGTTTCTGGATGTGTTCTAATGGGTCTTTGCAGTGCAGAATATGCATCTGAgattaggatttcagaaatataaggaaaaacacccttgtttctact-3'
WSN HA with coding sequence in caps: 3'-tcgttttcgtccccttttatttttgttggtttTACTTCCGTTTTGATGACCAGGACAATATACGTAAACATCGATGTCTACGTCTGTGTTATACATATCCGATGGTACGCTTGTTGAGTTGGCTGTGACAACTGTGTTATGAGCTCTTCTTACACCGTCACTGTGTAAGACAATTGGACGAGCTTCTGTCGGTGTTGCCCTTTGATACATTTAATTTTCCTTATCGGGGTGATGTTAACCCCTTTACATTGTAGTGGCCTACCGAGAACCCTTTAGGTCTTACGCTGAGTGACGAAGGTCGCTCTAGTACCAGGATGTAACATCTTTGTGGTTTGAGACTCTTACCTCGTACAATAGGTCCTCTAGAGTAGCTGATACTCCTTGACTCCCTCGTTAACTCGAGTCATAGTAGTAATCTTTCTAAGCTTTATAAAGGGTTCCTTTCAAGTACCGGGTTGGTGTGTAAGTTGCCTCATTGTCATAGTACGAGGGTATCCCCTTTTTCGTCAAAAATGTCTTTAAACGATACCGACTGCTTCTTCCCCCTAAGTATGGGTTTCGACTGGTTAAGGATACACTTGTTATTTCCCTTTCTTCAGGAACATGATACCCCACAAGTAGTGGGCAGATCGTCACTACTCGTTGTCTCAGAGATATCATTACCTTTACGAATACAGAGACATCGCAGAAGTTTAATATTGTCCTCTAAGTGGGGCCTTTATCGACGTTCCGGGTTTCATTCTCTAGTTGTACCCTCCTACTTGATAATGACCTGGAACGATCTTGGGCCTCTGTGTTATTATAAACTCCGTTGACCATTAGATTATCGTGGTACCATACGAAAGCGTGACTCATCTCCCAAACTCAGGCCGTAGTAGTGGAGTTTGCGCAGTTACGTACTCACATTGTGCTTCACAGTTTGTGGGGTCCCTCGATATTTGTCGTTAGAGGGAAAGGTCTTATATGTGGGTCAGTGTTATCCTCTCACGGGTTTTATACAGTCCTCATGGTTTAACTCCTACCAATGTCCTGATTCTTTGTAGGGTAGGTAAGTTATGTCTCCAGATAAACCTCGGTAACGACCAAAATAACTCCCCCCTACCTGACCTTACTATCTACCTACCATACCAATAGTAGTAGTCTTACTTGTCCCTAGTCCGATACGTCGCCTAGTTTTTTCGTGTGTTTTACGGTAATTGCCCTAATGTTTGTTCCACTTGAGACAATAGCTCTTTTACTTGTGAGTTAAGTGTCGACACCCATTTCTTAAGTTGTTGAATCTTTTTTCCTACCTTTTAAATTTATTTTTTCAACTACTACCCAAAGACCTGTAAACCTGTATATTACGTCTTAACAATCAAGATGACCTTTTACTTTCCTGAAACCTAAAGGTACTGAATTTACACTTCTTAGACATGCTCTTTCATTTTTCGGTTAATTTCTTATTACGGTTTCTTTAGCCTTTACCCACAAAACTCAAGATGGTGTTCACACTGTTACTTACGTACCTTTCACATTCTTTACCCTGAATACTAATAGGTTTTATAAGTCTTCTTAGTTTCAACTTGTCCCTTTTCTATCTACCTCACTTTAACCTTAGTTACCCCCACATAGTCTAAGACCGCTAGATGAGTTGACAGCGGTCAAGTGACCACGAAAACCAGAGGGACCCCCGTTAGTCAAAGACCTACACAAGATTACCCAGAAACGTCACGTCTTATACGTAGACTctaatcctaaagtctttatattcctttttgtgggaacaaagatga-5'
Rnd1rev426: 3'-gtgtgtaagttgcctcattgtcatagtacNNNNNNNNTCTAGCCTTCTCGTGTGCAGACTTGAGG-5' Rnd1rev849: 3'-agtttgcgcagttacgtactcacNNNNNNNNTCTAGCCTTCTCGTGTGCAGACTTGAGG-5' Rnd1rev1275: 3'-actactacccaaagacctgtaaaNNNNNNNNTCTAGCCTTCTCGTGTGCAGACTTGAGG-5' Rnd1rev1698: 3'-ctaatcctaaagtctttatattcctttttgtgggaaNNNNNNNNTCTAGCCTTCTCGTGTGCAGACTTGAGG-5'
Briefly, the steps are as follows:
The library of HA genes is subjected to PCR with primers to create subamplicons of \(\approx 425\) internal nucleotides (or less) as well as additional adaptor and primer-binding sequences. In the example above, PCRs are performed with the following primer pairs:
Rnd1for1andRnd1rev426: these primers create a subamplicon spanning nucleotides 1 to 426 (codons 1 to 142) of the WSN HA, which is the part of the sequence shown in caps above.
Rnd1for427andRnd1rev849: these primers create a subamplicon spanning nucleotides 427 to 849 (codons 143 to 283).
Rnd1for850andRnd1rev1275: these primers create a subamplicon spanning nucleotides 850 to 1275 (codons 284 to 425).
Rnd1for1276andRnd1rev1698: these primers create a subamplicon spanning nucleotides 1276 to 1698 (codons 426 to 566).Note that each of these subamplicons begins and ends on a full codon. This is important; design your primers to do the same.
The subamplicons are diluted so that the total number of ssDNA molecules is less than the sequencing depth. Since each subamplicon molecule has 8
Nnucleotides at each end, there are \(4^{16} \approx 4.3 \times 10^9\) unique barcodes. Since the number of unique barcodes is much greater than the total number of molecules in the dilution, each barcode will generally be associated with just one molecule.The barcoded subamplicons are further amplified by primers that add the Illumina adaptors, and perhaps Illumina indices for multiplexing. For instance, here are two example primers for this second round of PCR:
>Rnd2forUniversal: forward primer for the round 2 PCRs that adds the Illumina adaptor AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCC >Rnd2revIndex: reverse primer for the round 2 PCRs that adds the TruSeq index 1 (in lower case) as well as the Illumina adaptor CAAGCAGAAGACGGCATACGAGATcgtgatGTGACTGGAGTTCAGACGTGTGCTCTTCCThe products of the round 2 PCRs are sequenced with overlapping paired-end reads of sufficient length to cover the entire subamplicon, and the sequencing data are analyzed with the standard Illumina pipeline to generate FASTQ files with the R1 and R2 reads.
The FASTQ files are analyzed with dms2_batch_bcsubamp. For the example shown above, the
--alignspecsparameter would be1,426,36,38 427,849,32,32 850,1275,31,37 1276,1698,46,45.
Some important considerations if you are designing such an experiment:
Make sure that each primer begins / ends on a full codon – you do not want a codon split among subamplicons.
Make sure that your subamplicons are not excessively long, as the Illumina sequencing quality falls off near the end of reads. Although the example above uses four subamplicons to sequencing the 1.7 kb HA gene, in the actual experiments of Doud and Bloom (2016) on this gene, they instead chose to use six subamplicons so that they did not need high quality at the end of the reads.
Programs and examples¶
The dms_tools2 software contains programs to analyze the data generated by barcoded-subamplicon sequencing:
dms2_bcsubamp processes the FASTQ data to count mutations.
dms2_batch_bcsubamp runs dms2_bcsubamp on a set of samples and creates summary plots. In general, you will have an easier time if you use dms2_batch_bcsubamp rather than runnning dms2_bcsubamp directly.
Here are some example analyses:
More details and notes¶
The Bloom lab has now published a number of papers using this deep sequencing strategy. The methods sections of those papers have more details. For instance, see:
If you want really detailed information, you can look at some lab notes. Here are Jesse Bloom’s lab notes from an early iteration of this strategy on influenza HA, and here are Mike Doud’s lab notes from an experiment similar to that described in Doud et al (2017).


